DFT Calculations of Charge Transfer Parameters
The performance of devices such as organic light-emitting diodes (OLEDs) and organic thin-film solar cells is strongly influenced by the carrier mobility of the organic semiconductor materials they employ. To theoretically predict carrier mobility, it is essential to accurately determine key charge transfer parameters, including transfer integrals and site energies, based on the electronic states of the constituent organic molecules.
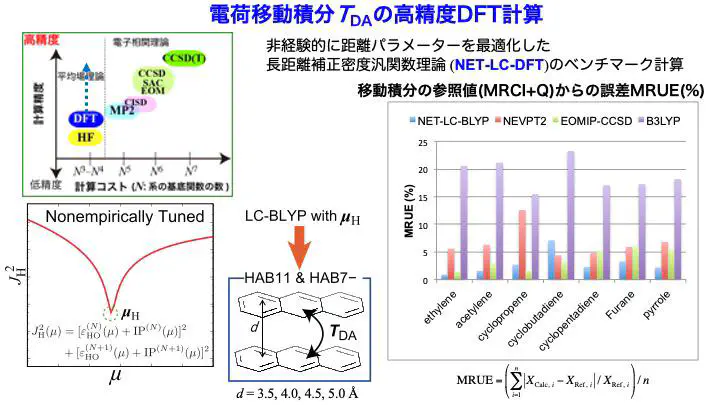
In this work, we propose that optimizing the range-separation parameters in long-range corrected (LC) density functional theory (DFT) for the individual molecules making up the organic semiconductors makes it possible to efficiently and reliably compute intermolecular electron and hole transfer integrals as orbital interactions within the DFT framework.
We validated this approach through benchmark calculations using the HAB11 and HAB7- datasets for hole and electron transfer integrals. Our results demonstrated that the proposed method achieves accuracy comparable to — and in some cases exceeding — that of computationally demanding coupled-cluster and multireference perturbation theory methods while significantly more efficient.