電荷移動パラメータのDFT計算
有機ELや有機薄膜太陽電池などのデバイスの効率は、その有機半導体が持つキャリア移動度に大きく依存します。 キャリア移動度を理論から見積もるには、移動積分やサイトエネルギーなどの主要な電荷移動パラメータを、 構成有機分子の電子状態から決定する必要があります。
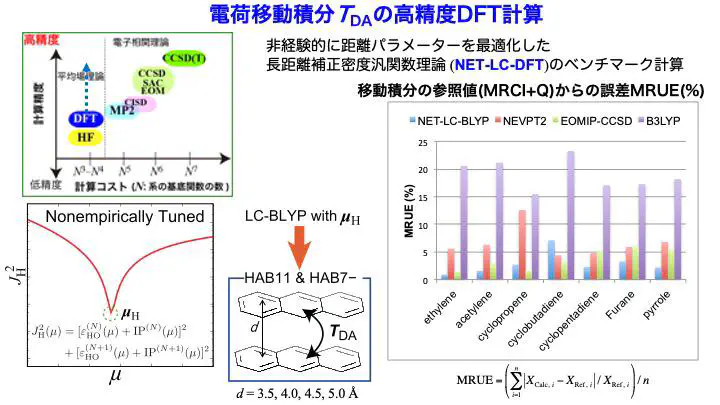
我々は、長距離補正(LC-)密度汎関数理論(DFT)の距離分割パラメータを、 有機半導体の構成分子に対して非経験的に最適化することで、 分子間の電子・ホール移動積分をDFTの軌道相互作用として、 高精度かつ効率的に求めることができることを提案しています。
ホール/電子 移動積分データセットであるHAB11とHAB7-に対してベンチマーク計算を行ったところ、 非常に時間がかかる結合クラスター法や多参照摂動理論法を用いた結果より、 我々が提案した手法が良い精度の結果になることを報告しました。
(2019).
Calculation of Charge-Transfer Electronic Coupling with Nonempirically Tuned Range-Separated Density Functional.
The Journal of Physical Chemistry C Vol. 123, num.18, pp. 11351–11361 (2019).
(2015).
Charge-transfer matrix elements by FMO-LCMO approach: Hole transfer in DNA with parameter tuned range-separated DFT.
Chemical Physics Letters Vol. 621, pp.96–101 (2015).