Research
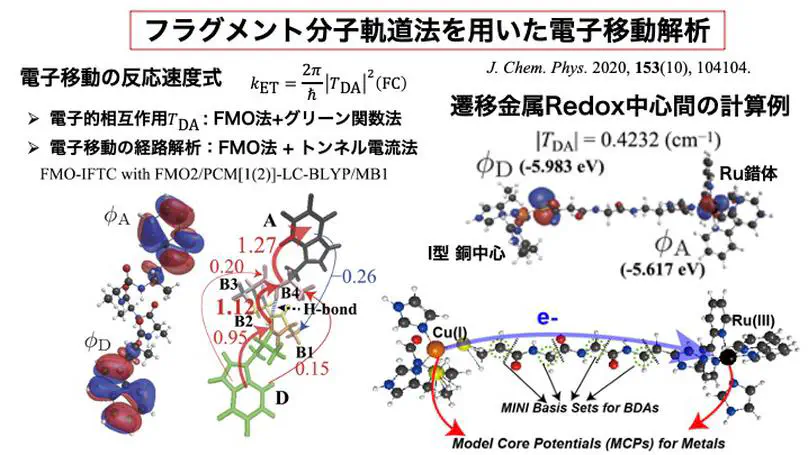
We develop novel theoretical methods based primarily on quantum chemical calculations, implementing them into computational programs to investigate the microscopic mechanisms of chemical reactions where quantum effects play a crucial role — including electron transfer, excitation energy transfer, and proton transfer — in organic materials, solutions, and biological systems.
Our ultimate goal is to theoretically design and discover highly functional molecular materials by gaining a deeper understanding and control of these fundamental reaction processes.