First-principles Calculations of Electron Tunneling Pathways in Proteins
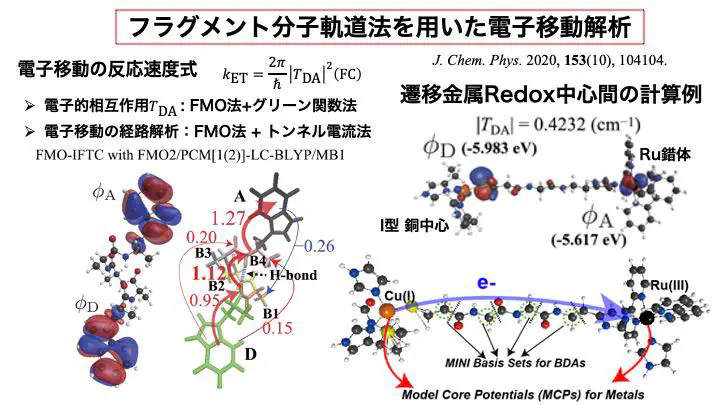
Electron transfer (ET) reactions in proteins are central to biological energy conversion processes, such as photosynthesis and cellular respiration. Cofactors such as chlorophyll, quinone, heme, and iron-sulfur clusters act as redox centers in these reactions, facilitating electron transfer. Although proteins behave as insulators, electrons can move between these spatially separated cofactors (typically over distances greater than 5 Å) via quantum tunneling.
A fundamental, unresolved question is whether proteins have developed specific structural features through evolution that create optimized electron tunneling pathways, thereby promoting efficient biological electron transfer reactions. We conduct first-principles investigations using quantum chemical calculations on proteins to address this question.
Given the large size of protein molecules, directly computing their electronic states is computationally demanding. To overcome this, we have developed an electron tunneling pathway analysis method based on the Fragment Molecular Orbital (FMO) method. This approach divides large protein structures into smaller fragments, approximating the electronic state of the entire protein from the electronic states of its fragments. By leveraging the spatial localization of fragment electronic states, this method enables efficient identification of the specific fragments through which tunneling electrons move, thus making pathway analysis feasible while avoiding prohibitive computational costs.
At present, our method integrates the FMO framework with Density Functional Theory (DFT), Model Core Potentials (MCP), and the Polarizable Continuum Model (PCM), allowing us to incorporate electron correlation, metal center effects, and solvent effects into the analysis of electron tunneling pathways in proteins.