Charge Transfer Simulation in Organic Semiconductors
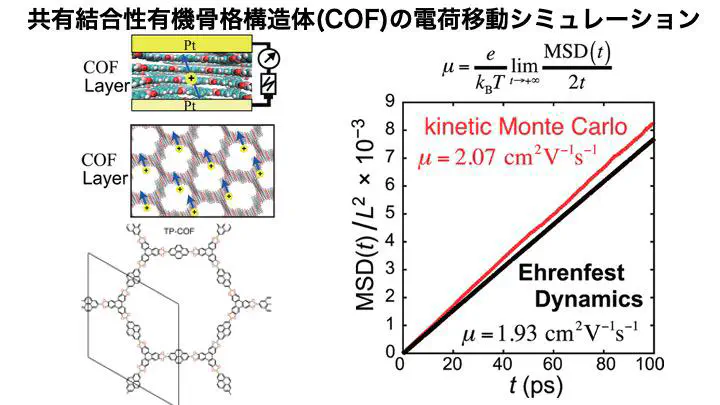
Covalent Organic Frameworks (COFs) are porous crystalline materials constructed from organic molecules, known for their remarkable properties such as self-assembly, lightweight, thermal stability, and gas adsorption and storage capabilities. In addition to these features, several COFs exhibit electrical conductivity, with some achieving carrier mobilities exceeding 1 cm$^2$ V$^{-1}$ s^{-1}.
Our research focused on TP-COF, the first reported COF with p-type semiconductivity, where we elucidated its charge transport mechanism using multiscale multiphysics simulations of charge transfer processes. This study also developed a new computational approach to efficiently evaluate key charge transfer parameters — including site energies and charge transfer integrals — by integrating the empirical Density Functional Tight-Binding (DFTB) method with the Fragment Molecular Orbital (FMO) method.
Additionally, we applied this methodology to investigate a molecular amorphous material designed as an alternative to spiro-MeTAD, a commonly used hole-transport material in perovskite solar cells.